海河文库|FDA QMSR新规2026年2月2日正式生效,QSIT将正式退出历史舞台
2026年2月2日,美国食品药品监督管理局FDA QMSR,21CFR Part 820修订版将正式强制执行。这一里程碑式的监管变革,标志着美国医疗器械质量管理体系全面接轨国际标准,通过引入ISO 13485:2016 核心要求、优化检查流程、扩大记录审查范围,为全球医疗器械企业进入美国市场构建更协同、更高效的合规框架,同时进一步强化对患者安全的保障。
新规核心变革:从 “QS” 到 “QMSR” 的重大调整
1,法规框架重构,国际标准深度融合
此次修订将QSR《质量体系法规》正式更名为QMSR《质量管理体系法规》,核心亮点是通过 “引用并入” 方式,将国际标准化组织(ISO)发布的ISO 13485:2016《医疗器械质量管理体系用于法规的要求》及ISO 9000:2015 Clause 3《质量管理体系基础和术语》 全面纳入FDA 监管要求。QMSR《质量管理体系法规》除了引用 ISO 13485 和 ISO 9000 第 3 条之外,还制定了额外的要求和规定,以澄清 ISO 13485中使用的某些预期和概念。这些补充确保引用 ISO 13485 不会与其他适用的 FDA 要求产生冲突。FDA 指出,ISO 13485:2016 要求与旧版 QS 法规 “实质相似”,可确保企业质量管理体系对 “安全有效医疗器械持续生产” 的保障水平,同时大幅降低跨国企业的合规成本,实现与全球主要监管机构的标准协同。
2,检查体系全面升级,QSIT 正式退出历史舞台
2026年2月2日起,FDA将停用沿用多年的QSIT《质量体系检查技术》,启用全新的《医疗器械制造商检查合规程序》(7382.850)。新的检查流程将始终关注风险,在检查过程中,对一项要求的评估可能需要对 QMS 中其他领域的其他要求进行评估。新的基于风险的检查流程与QMSR《质量管理体系法规》 相一致,重点关注对患者和/或用户的风险。为了便于关注风险并反映 QMS 流程之间的关系,新的基于风险的检查流程将QMSR要求组织成 6 个 QMS 领域和 4 个其他适用的 FDA 要求 (OAFR)。
6个QMS 领域和4个其他适用的 FDA 要求 (OAFR):

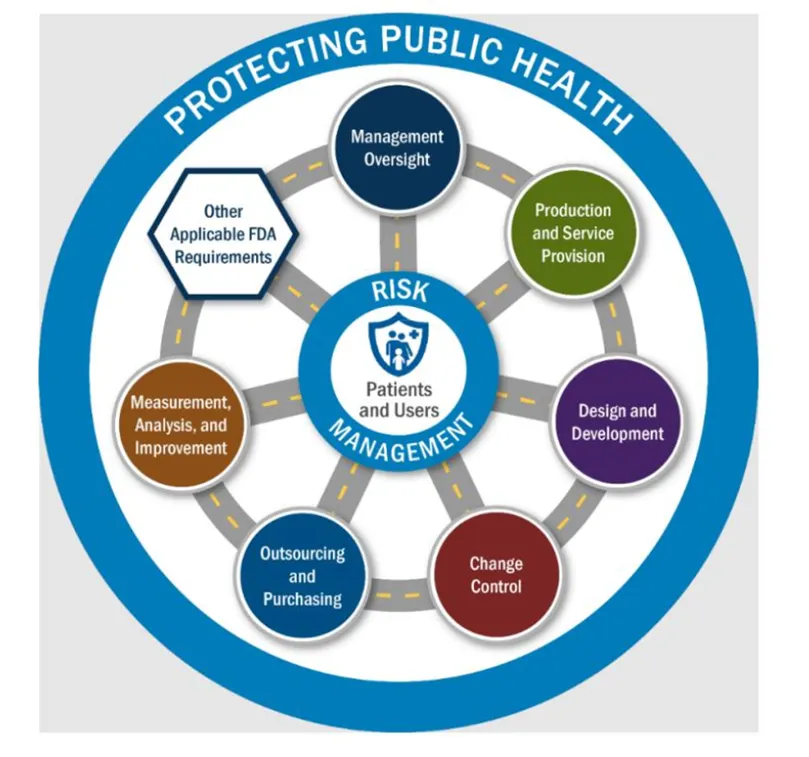
患者和用户是美国食品药品监督管理局(FDA)医疗器械检查工作的核心关注对象,他们在上图中处于中心位置。围绕患者和用户的风险管理圆圈体现了 FDA 对利用制造商的风险管理文件来帮助将检查重点放在风险上的重视。QMSR 要求被表示为六个圆圈(即QMS区域)以及一个六边形,代表另外四项(4)适用的 FDA 要求(OAFRs)。连接各个 QMS 区域和OAFRs 的“道路”反映了它们之间的联系以及检查过程的灵活性。外圈说明了检查过程有助于实现 FDA 保护公众健康的使命。
3,记录审查范围扩大
QMSR赋予FDA检查管理评审、质量审核和供应商审核报告的权力。
在2026年2月2日的QMSR生效日当天或之后,美国食品药品监督管理局FDA将检查管理评审、质量审核和供应商审核。原QS法规第820.180(c)条中存在的例外情况,在QMSR中未被保留。正如最终规则的前言所规定的,由于制造商被要求向其他监管机构提供此类文件,因此向FDA检查员提供这些记录不会给他们带来额外负担。这些记录在正常业务过程中保存,应在检查时随时可用。
海河生物提供专业服务与支持
海河生物深耕生物医药法规和检测行业20年,始终密切关注 FDA 最新监管要求,已成功为500余家企业提供了质量体系咨询服务,诸多企业更是以“无缺陷”的结果通过工厂检查。我们熟悉并了解美国FDA的现场审核要求,擅长根据客户现有的质量管理体系进行提升和完善,协助客户以最小的变动满足各国监管要求,为客户构建真实有效可落地的质量管理体系解决方案。如果您对 FDA 器械质量管理体系 QMSR 有任何疑问,欢迎联系和咨询(sales@harbios.com),我们将竭诚为您服务。
🎯 我们的目标:助您在复杂监管环境中保持领先,确保产品合规、顺利上市。
如需转载,请标明来自海河生物
关于海河生物
海河生物是一家专业为医疗器械和药品相关的研究机构、研发和生产企业及相关监管部门提供产品全生命周期服务的平台性公司,在生物医药CRO领域具有极高的行业地位。海河生物旗下业务涵盖产品全生命周期服务、服务型制造、智能制造和第三方检测认证,与我国“十四五规划”高度契合。
海河标测:
从事医疗器械、药品、药械结合产品和生物医药环境标准化和定制检测和监测服务,海河生物的医疗器械和药品检验检测业务,尤其是医疗器械检测具有国内、国际先进检测标准相关资质,可以做到一次检测满足不同地区和市场的检测要求。
海河咨询:
向客户提供中国、美国、欧盟、巴西、加拿大和东南亚国家等国家地区的医疗器械、药品和药械结合产品全生命周期法规政策咨询、注册认证辅导、上市后不良事件处理、技术转化、培训等相关服务。
海河CDMO:
海河生物的医疗器械委托研发生产平台,是按照国家药监局医疗器械生产质量管理规范、美国药监局 QSR 和 ISO13485 要求建立的为器械产学研、医工转化和初创型团队服务的平台,承接医疗器械产品定制、外包研发、外包生产的业务,生产能力涵盖 IVD 诊断试剂,IVD 诊断设备,医疗设备、外科高值耗材等产品 。

扫码关注我们
让生物医药产品无国界